Tagged under:

The pharmacokinetic behaviour of compounds is linked to their efficacy and thus is critical for drug discovery. Understanding how to optimise compounds according to multiple simultaneous criteria is a great advantage in focusing design efforts.VolSurf+ creates 128 molecular descriptors from 3D Molecular Interaction Fields (MIFs) produced by our software GRID, which are particularly relevant to ADME prediction and are also simple to interpret. One example would be the interaction energy moment descriptor between hydrophobic and hydrophilic regions, which is important for membrane permeability prediction. These can then be used with provided chemometric tools to build statistical models.VolSurf+ also comes with a number of models that we have developed using both public and pharmaceutical data, including passive intestinal absorption, blood-brain barrier permeation, solubility, protein binding, volume of distribution, and metabolic stability.A more complete description of the original VolSurf method and derived models is available in the references section.
Key Features:-
- Calculate ADME relevant descriptors and perform statistical modelling using experimental data
- Predict the behaviour of new compounds based on new or established ADME models
- Interactively sketch structural modifications to optimise ideas according to multiple criteria
- Select compounds with similar ADME properties to a query structure
Homepage - http://www.moldiscovery.com/soft_vsplus.php
Download
Tagged under:
Programme GRID is a computational procedure for determining energetically favourable binding sites on molecules of known structure. It may be used to study individual molecules such as drugs, molecular arrays such as membranes or crystals, and macromolecules such as proteins, nucleic acids, glycoproteins or polysaccharides. Several different molecules can be processed one after the other.
The best-known application of GRID in Structure-Based Drug Design [ Nature 363, 418-423 (1993) ] shows the potentiality of the approach. Potent inhibitors based on the crystal structure of influence virus sialidase were designed, leading to the recently approved drug RELENZA® from Glaxo.
Moreover GRID can be used to understand the structural differences related to enzyme selectivity, a fundamental field in the rational design of drugs [ J.Med.Chem. 43, 3033-3044 (2000) ].
GRID maps can also be used as descriptors input in statistical procedures like CoMFA, GOLPE or SIMCA for QSAR or 3D-QSAR analyses [ J.Med.Chem. 40, 4089-4102 (1997) ]. Successful applications of GRID are reported in the field ADME and metabolism through VolSurf program and MetaSite program.GRID may be used after a novel ligand has been designed and fitted to its receptor. The GRID map is then searched for places at which water molecules have NOT been displaced by the ligand from the site. These remaining waters should all be making appropriate hydrogen-bonding contacts to the atoms of the ligand, or to the receptor, or to neighbouring water molecules.
However, it often happens that water molecules could be trapped in hydrophobic regions between the ligand and receptor molecules, at places where they cannot make appropriate hydrogen bonds. It is not easy to detect this possibility without using GRID. These trapped waters are unfavourable and tend to destabilise ligand binding. The design of the ligand molecule should therefore be modified, so that the unfavourably trapped water molecules will be displaced when the new modified ligand binds.
Homepage - http://www.moldiscovery.com/soft_grid.php
Download
Tagged under:

The GOLD Suite consists of the following packages: Hermes for 3D visualisation pre- and post-docking, interactive docking setup.Hermes is a 3D visualiser with particular emphasis on functionality for the analysis of protein-ligand interactions.GOLD for protein-ligand docking:GOLD is a program for calculating the docking modes of small molecules in protein binding sites and is provided as part of the GOLD Suite, a package of programs for structure visualisation and manipulation (Hermes), for protein-ligand docking (GOLD) and for post-processing (GoldMine) and visualisation of docking results. Hermes acts as a hub for many of CCDCs products, for more information please refer to the Hermes product page.GoldMine for post-processing of docking results:GoldMine is a tool for post-processing of GOLD docking results. GoldMine can be used to combine and analyse several docking runs, e.g. docking runs carried out against different protein models may be combined within a GoldMine database and analysed for selectivity and specificity.Docking runs carried out using one protein model but scored using different scoring functions may also be combined and evaluated within a GoldMine database. GoldMine is provided as part of the GOLD Suite, a package of programs for structure visualisation and manipulation (Hermes), for protein-ligand docking (GOLD) and for post- processing (GoldMine) and visualisation of docking results.
GOLD features include:
A genetic algorithm (GA) for protein-ligand docking
An easy to use interface with interactive docking set-up via Hermes
A comprehensive docking set-up wizard
Full ligand flexibility
Partial protein flexibility, including protein side chain and backbone flexibility for up to ten user-defined residues
Energy functions partly based on conformational and non-bonded contact information from the CSD
A variety of constraint options
Improved flexible ring handling
Automatic consideration of cavity bound water molecules
Improved handling and control of metal coordination geometries
Improved parameterisation for kinases and heme-containing proteins
Automatic derivation of GA settings for particular ligands
A choice of GoldScore, ChemScore, Astex Statistical Potential (ASP) or Piecewise Linear Potential (PLP) scoring functions
Extensive options for customising or implementing new scoring functions through a Scoring Function Application Programming Interface, allowing users to modify the GOLD scoring-function mechanism in order to either: implement their own scoring function or enhance existing scoring functions; customise docking output
A ChemScore Receptor Depth Scaling (RDS) rescore option so that the score attributed to hydrogen bonds is scaled depending on the depth in the binding pocket
Automatic rescoring with an alternate scoring function at the end of a docking run.
General Hermes features include:
The ability to view protein and ligand structures from external files
Easy navigation of the protein structure and control of what is displayed
A selection of visualisation options including a choice of display styles, colours, labelling schemes, and the ability to hide and then re-display atoms, residues, ligands, water molecules, etc
Functionality to generate auxiliary objects such as centroids and ribbons
The ability to restrict the contents of the rendered image based on the Z coordinate of individual objects (i.e. apply Z-clipping)
The ability to measure and display distances, angles and torsion angles
The option to find and display hydrogen bonds and non-bonded clashes, and to customise how they are defined geometrically
Various display output options (e.g. png, jpg, bmp)
The ability to edit ligands either manually or automatically to Cambridge Structural Database (CSD) conventions
Stereoviewing functionality
The ability to define and calculate descriptors
Links to a pre-computed library of ligand geometries (Mogul)*
Links to a precomputed library of non-bonded interactions (IsoStar)*
* CSD System subscribers only
Download :
http://www.easy-share.com/1910194212/C.G.S.4.12.rar
Mirror link:
http://hotfile.com/dl/42339652/f592887/C.G.S.4.12.rar.html
Mirror link:
http://sharingmatrix.com/file/4748544/C.G.S.4.12.rar
Mirror link:
http://turbobit.net/23u9epn38ug2.html C.G.S.4.12.rar
Mirror link:
http://ugotfile.com/file/1323105/C.G.S.4.12.rar
Mirror link:
http://uploading.com/files/89cd11ae/C.G.S.4.12.rar/
Tagged under:

Diamond is our outstanding molecular and crystal structure visualization software. It integrates a multitude of functions, which overcome the work with crystal structure data - in research and education as well as for publications and presentations. Diamond does not only draw nice pictures of molecular and crystal structures like most of its competitive programs do. It offers an extensive set of functions that let you easily model any arbitrary portion of a crystal structure from a basic set of structural parameters (cell, space group, atomic positions). With its high data capacity, its wide range of functions beginning with the generation of molecules reaching up to the construction of rather complicated inorganic structural frameworks, Diamond is a comprehensive tool for both molecular and solid state chemists as well as for surface and material scientists.
Features of Diamond Version 3
The following gives you an overview of the new features of version 3, not including all the features that are already available in the previous version 2.
Multiple structure data sets, multiple pictures
The new Diamond 3 document format enables storage of multiple structure data sets as well as multiple structure pictures. New desktop elements are:
the table of data sets,
an overview of pictures (as thumbnails) associated with a dataset or with the whole document,
the navigation tree,
several new views (powder pattern, distances and angles, properties, etc.).
Powder pattern simulation
Powder diffraction data can now be calculated and displayed in both table and diagram. Table and diagram update automatically when structure parameters change.
Properties pane and more auxiliary views
The new Properties pane shows informations in context with the current situation, e.g. properties of the selected atom, distances around the selected atom, centroid or best plane or line through selected atoms, and many more.
Distances and angles table now provide histograms and statistics.
The data brief for a short overview of structure parameter data.
The Info Tip window gives you information about the object under the mouse cursor.
Individual assignment of model
In Diamond 2, the model (ball-and-stick, space-filling etc.) was a global setting. Now it can be assigned individually to selected atoms. That means you can display ball-and-stick, space-filling, ellipsoid, and sticks or wires in one and the same picture.
Create POV-Ray scenes
The POV-Ray assistant helps you to create photo-realistic pictures for this outstanding, free available ray-tracer, with features like textures, background, ray-tracing, multiple light sources, etc.
Geometry enhancements: Planes, lines and more
Planes can now be defined by hkl or as (best) plane through a set of three or more atoms (or a line through two or more atoms). The planarity (linearity) can be checked.
Besides this, distances of atoms from planes or lines as well as angles between planes and/or lines can be calculated or measured interactively.
More features and enhancements
Assistant for creation of animation scenes,
walk-through mode,
composition of pictures from different data sets,
support of "molecular data" (structure data with no cell and symmetry),
a step-by-step assistant for users who are not so familiar with Diamond,
an automatic picture creation,
cut/copy/paste between structures and pictures,
printout of multiple pictures per page,
and more.
System Requirements
Personal computer with MS Windows® 98, ME, 2000, XP, or Vista (NT® 4.0 on request)
Microsoft Internet Explorer 5.01 (or higher)
Pentium® II compatible processor (or higher)
64 MByte of RAM (or more)
Graphics resolution of 1024x768 and 16 bit color depth (or higher)
CD-ROM drive
Hard disk with minimum 100 MB free disk space (or more)
Microsoft compatible mouse
Homepage:- http://www.crystalimpact.com/diamond/
Download
Tagged under:

Avogadro is an advanced molecular editor designed for cross-platform use in computational chemistry, to molecular modeling, bioinformatics, materials science and related fields.
It offers a flexible framework and a powerful rendering plug-in architecture. After installing the program stpnovitsya portable. Runs through avogadro.exe, located in the folder [bin]
Osban
* Cross-platform: Molecular Designer for Windows, Linux and Mac
* Free, Open Source. Easy to install and available all the source code (GNU GPL)
* International: Translations into Chinese, French, German, Italian, Russian, Spanish and more
* Intuitive: built-in easy to work for students and researchers as advanced.
* Speed: Support multithreading visualization and computation.
* Expandable: Plugin architecture for developers, including interactive instrumentoy, commands and scripts Python
* Flexible: Features include import of chemical File Open Babel, a computer package, crystals, biomolecules, etc.
Features:
* Built-in molecular mechanics (including MMFF94 and UFF).
* Enter the generation of Gaussian, GAMESS-US, Molpro, NWChem, Q-Chem, MOPAC, with the addition of packages in the future.
* Support crystallographic cells.
* Developer of oligopeptides.
* Visualizing isosurfaces and orbits, including the Gaussian cubes, OpenDX, and Gaussian files fchk.
* Animation calculated molecular vibrations and the trajectories of the reaction.
* Full range of views with zoom and continues to expand.
* Export models in POV-Ray.
In addition, Avogadro provides libraries (libavogadro), which can be used for software development.
The program does not provide the choice of language. Version 1.0.1 has only in English. It is proposed to fully russifitsirovanaya version 1.0.0 of 2009-10-23.
Avogadro 1.0.1 was released on 28 April 2010.
What's New
This is a stable bug-fix release, recommended for all users. The following list includes some of the bugs fixed since Avogadro 1.0.0:
* Fixed crashes and highlighting in Properties extension
* Fixed crash on exit if forcefields are missing
* 3D structures are now downloaded when using Network Fetch
* Fixed crash in Project Tree Editor
* Fixed open and save on Windows if non-latin symbols occur in path
* Fixed improper behavior of PluginListView (plugin info didn't change on keyboard movements)
New supplementary files:
* New avopkg tool for management of 3rd party plugins
* File avogadro.prf for integration with QMake build system for 3rd party extension developers
* Man pages for Unix platforms
Update: Apr 28, 2010
Platforms: Windows NT/2000/XP/2003/Vista/7
Languages: English, Russian
Download
Tagged under:

Chemissian is an analyzing tool of molecules electronic structure and spectra. It can manipulate molecular orbital energy-level diagrams (Hartree-Fock and Kohn-Sham orbitals), calculated and experimental UV-VIS electronic spectra, electronic/spin density maps and prepare them for publication. Chemissian has a user-friendly graphical interface and lets you examine and visualize data from the output of Gaussian, US-Gamess, Firefly/PC-Gamess quantum chemical program packages. Chemissian tools helps you to investigate nature of transitions in UV-vis spectra, bonding nature, etc.
Chemissian Features:
Build Molecular Orbitals energy level diagrams
• Due to the integrated graphical editor it is easy to add text labels to the diagrams, make connector lines between MO energy levels, text labels, occupy the energy levels with the electrons.
• You can analyze the electronic structure of molecules: you can move between energy levels simply using the keyboard cursor buttons and in a useful way obtain information about contributions to the current molecular orbital from atoms or molecular fragments and present the data in the most useful and demonstrative way: on the contribution diagram or directly on the MOs themselves
Build, visualize and interpret UV-Visible Spectra from Gamess, Firefly and Gaussian outputs
Chemissian with its exciting and unsurpassed graphical analyzer of properties and composition of MOs, has the wide range of capabilities for analysis of electronic spectra of molecules. Chemissian offer tools for building electronic UV/VIS spectra directly from quantum-chemical data from GAMESS, Firefly(PC-GAMESS) or GAUSSIAN output files:
• Build spectrum in one step: just load Gamess/Firefly/Gaussian outputs with TDDFT/CIS data.
• Having experimental spectra you can compare it with the calculated ones on the single diagram in the same wavelength scale.
• Any number of spectra may be added on single diagram, which is useful, e.g. when solvent influence on the spectrum is considered.
• Like in MOs editor it is possible to move between spectra peaks and correlate the current peak with transitions between MO energy levels.
• Chemissian allows editing the obtained spectrum diagram by adding text labels and other graphical objects. Different energy units are available.
• Using Chemissian tools it is easy to investigate the nature of spectra transitions, e.g. metal-to-ligand charge transfer, ligand-to-ligand charge transfer, pi-pi*, etc. based on information about the compositions of molecular orbitals from output files of gamess/Firefly/gaussian.
Analyze electronic density distribution
• Using Chemissian you can analyze electronic and spin density distribution, difference (also called "defomation") density, individual molecular orbital, and arbitrary linear combination of them (e.g. for plotting Fukui functions).
• Chemissian can build the distributions as two-dimensional contour maps or
• Build distribution along the given line (one-dimensional).
• To build densities only standart gamess/Firefly/gaussian output file is used, e.g. no cube-files are needed.
Calculate populations and valences
• Chemissian can calculate Mulliken and Simple populations of AOs, Shells, "Spherical Harmonics", Atoms or molecular fragments (any group of atoms).
• Also you can choose to calculate valences of AOs, Shells,"Spherical Harmonics", Atoms and fragments.
• Analyze molecular orbital composition - calculate contributions from atomic orbitals, atoms, molecular fragments, shells, etc. to the MOs.
Calculate quantum-chemical bond order indexes and overlap populations
Use Chemissian to investigate bonding nature in the molecules - calculate quantum-chemical bond order indexes and overlap populations for every bond in molecule. You can also analyze "generalized bond, e.g. "bond" between molecular fragments.
Work with several calculations at the same time
In a single document accumulate and analyze results of several calculations, e.g. load several GAMESS/Firefly/Gaussian ouput files. Simple example: having you the source reagents and the final reaction product you want to understand the changes that have occurred on the electronic structure level - you may add several calculations (reagent and product) at the same diagram, and they will be presented in the common energy scale, you can switch between different calculations, compare and analyze electronic structures all the participants at the same time.
Save the results in a single file
Save the obtained document in a special file format, which allows to keep all data in a single compressed file (uncompressed wave function takes up a lot of disk space!); at any time you will be able to open and continue working with the saved document, analyze, edit the data, send it to your colleagues.
Download
Tagged under:
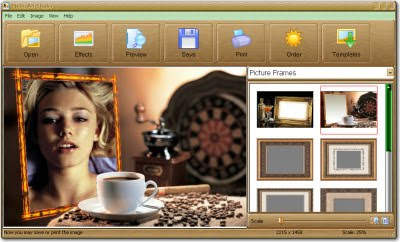
Photo Art Studio is a brand-new application for decorating photos and applying effects. Within literally a couple of seconds, you will be able to build up a stylish frame, create a postcard or a collage. The program includes a handful of functions necessary for processing digital photographs: automatic enhancement and editing, creating special effects, framing, adding decorations and labels. The distribution kit includes somewhat about three hundreds of decoration options, and this number increases in the full version!
Photo Art Studio features a pretty, customizable user interface, easy to get along with. The program is easy and comfortable to operate, and its capabilities exceed your expectations! After just a few minutes, you will master the art of professional decoration of photos of any kind. The built-in help system and the online tutorial will help you turn your pictures into true masterpieces!
So, go ahead and launch Photo Art Studio and then open the first photo in it. The editor pane will come up right next; there you can adjust the primary properties of the image and crop your image. Next, by choosing the operating mode, you can shape up the photograph with special filters, apply classical and modern frames, and create postcards and original collages.
Major Advantages of Photo Art Studio:
• Convenient user interface and ease of operation.
• Five automatic photo enhancement algorithms.
• Over three hundreds of decoration schemes: frames, masks, postcards, collages...
• Huge set of new templates in the full version, expandable template base.
Download links:
Download
Mirror
Tagged under:

Download
Search and Replace is an award winning, easy to use search and replace utility loved by programmers, webmasters, and novice computer users alike.
Search and Replace looks through multiple files for a string and can also replace it with another string. It can search subdirectories and ZIP files and do case sensitive or insensitive searches. Extensive support for grep style regular expression search & replacements includes operations that span more than one line, incrementing number replacements, and inserting the path & filename in replaced files. Binary search & replace is also available. You can specify multiple include/exclude file masks and filters based on file date & size. Control over replacements includes configurable replacement prompts and a display of replacements before they are made. An easily understood script editor makes frequent &/or complex multi-step search/replace operations easy to prepare. Advanced script operations include a boolean expression evaluator for added control over which files will be processed by the script. The internal context viewer allows you to edit text files. An "HTML Mode" makes plain text -> html character code substitutions. You can also "Touch" files (change time/date stamp). Search and Replace automatically detects if the file being searched is text and can be configured to launch with separate external editors for search hits in text vs. binary files. The program can also launch associated programs on "found" files, e.g., launch your web browser if the search hit occurs in an .htm or .html file.
If you need to find and replace text, or just find text, this utility is a must have. It is significantly faster than other Windows grep-only utilities and allows replacing for the same price as those programs which offer only grep capabilities.
Search and Replace is for Windows NT/2000/XP, Windows 2003, and Windows Vista. 32-bit and x64 versions are available.
Here are some of the basic features of Search and Replace. A history of features by version and an overview of proposed new features are also available.
+ Search and replace operations can be case sensitive or not, they can span multiple files via multiple file masks and filter settings, and they can span more than one line
+ The program can search-replace the Windows Clipboard. You can also do simple searches in the Search Results list (press F3) and you can also limit follow-up search &/or replace to only those files currently listed in the Search Results list.
+ Unicode files are detected automatically for search and replacing.
+ Scripts can be applied to Search/replace strings in the Binary Mode dialog. This allows special processing of RTF codes, HTML codes, Unicode files, with a lookup table created by the user along with several supplied by us.
+ Output in the Search and Replace "Results List" can be sent to your printer, saved to a file (your choice of delimiters), or viewed in your web browser via an automatically generated htm file. If you have a color printer you can print the html file to obtain a color coded output.
+ File operations can be carried out on the files in the search results list.
+ An HTML Mode that automatically makes plain text -> html special character substitutions during search &/or replace operations.
+ The internal "context" viewer allows you to view your search results in context with the surrounding text. You can jump from one "search hit" to the next and edit text files. Search and Replace automatically detects if the file being searched is text and, if not, will automatically view binary files using our freeware companion utility HexView. Alternatively, Search and Replace can be configured to launch with separate external editors for search hits in text vs. binary files. Links off the External Editors page provide information for using TextPad (Helios Software, www.textpad.com), Multi-Edit (American Cybernetics, www.multiedit.com), UltraEdit (IDM Computer Solutions, www.ultraedit.com), Hex Workshop (BreakPoint Software, www.bpsoft.com) and 'frhed' (Raihan Kibria, www.tu-darmstadt.de/~rkibria).
+ Search and Replace can also launch associated programs on "found" files, e.g., launch your web browser if the search hit occurs in an .htm or .html file.
+ Text files can be reformatted w/ word wrap at a specified column.
+ Text can be prepend or appended to a file.
+ An 'Ignore Whitespace' function can find a phrase regardless of where line breaks may lie.
+ The program can read the environment variables on the host computer and use them in a search/replace using the "%%envvar=" syntax. For example, the 'temp' environment variable can be referred using the string "%%envvar=temp%%".
+ Replacements, including those in script and binary operations, can be visualized in context before they are made.
+ You can (optionally) write files changed by a replacement operation to a backup directory and preserve original file time/date stamps during replacements.
+ Regular Expression operators include the basic grep-style substring and match operators, along with special replacement operators such as case change operators, file and path name replacements, counters, and binary mode operators. The Incrementing Counter operations let you allow you to quickly revise a sequence of numbers in one or more files or add numbers where no numbers exist originally. (See Regular Expressions English or Deutsch).
+ Scripts let you specify multiple search/replace combinations, save a particular search or replace string for later use, or process multiple drives/directories and/or file masks. Advanced Script Settings let you control program options, embed comments in scripts, and further specify the files processed during a script operation via a boolean expression evaluator. When building scripts you can easily add the the Search/Replace & Mask/Path values from the mail dialog by holding down the CTRL key while clicking on the Insert or Remove All buttons in the script editor. (See Script Editor page). Scripts include an iterative operator to repeat a script a specified number of times.Binary Mode search and replace lets you process binary files &/or text files with special characters such as tabs, line feed, carriage returns. (See Binary Editor Page).
+ Multiple include & exclude file masks let you, for example, search all ".htm" & ".html" files except the ".htm" files begin with "s" or "t". You can also create complex file masks to include some subdirectories while excluding others. Filter Options allow you, for example, to include only files created after a set date that are smaller than the size you specify.
+ Searching can be halted after the first hit in a file - useful for when you have to search large or many files. A 'no replace' flag is also available to turn off replacement functions - useful for administrators or users who need extra limits on the possibility of inadvertent replacements.
+ Scripts can be applied to the Search &/or Replace strings in advance of an operation. Activated via the Apply Script function, you can, for example, use a conversion script to deal with the plain text equivalents of html special characters or rtf 'escape' codes.
+ User control over replacement prompts/warnings includes one string at a time, one file at a time, all at once (no warnings), and combinations in between such as skipping all remaining occurrences in a file.
+ An enhanced "Touch" dialog allows you to change time/date stamp and attributes of files.
+ Search and Replace can be launched from Windows Start-Find Menu. Files &/or directories dropped from Explorer or File Manger into the Search and Replace main window are automatically recognized for processing. The Windows Explorer shell extension lets you launch Search and Replace on a specific file or group of selected files.
+ An 'UnDo' function is available to reverse the last search-replace (see F1 Hlp).
+ Search ZIP files without manually extracting files first.
+ User configurable fonts & colors.
+ Command line support (14 functions) includes control over most program options & ability to run search & replaces via batch files.
+ Help file includes many examples.
+ An established, mature program with a history of regular updates and solid support.
+ Much more...
Homepage http://www.searchandreplace.com